United States
Securities and Exchange Commission
Washington, D.C. 20549
FORM 10-Q
(Mark One)
| [X] | QUARTERLY REPORT PURSUANT TO SECTION 13 OR 15(d) OF THE SECURITIES EXCHANGE ACT OF 1934 |
For the quarterly period ended March 31, 2021
or
| [ ] | TRANSITION REPORT PURSUANT TO SECTION 13 OR 15(d) OF THE SECURITIES EXCHANGE ACT OF 1934 |
For the transition period from ____ to ____
Commission File Number 333-184948
Processa Pharmaceuticals, Inc.
(Exact name of registrant as specified in its charter)
| Delaware | 45-1539785 | |
(State or other jurisdiction of incorporation or organization) |
(IRS Employer Identification No.) |
7380 Coca Cola Drive, Suite 106,
Hanover, Maryland 21076
(443) 776-3133
Securities registered pursuant to Section 12(b) of the Exchange Act:
| Title of Each Class | Trading Symbol(s) | Name of each exchange on which registered | ||
| Common Stock, $0.0001 par value per share | PCSA | The Nasdaq Stock Market LLC |
Indicate by check mark whether the registrant (1) has filed all reports required to be filed by Section 13 or 15(d) of the Securities Exchange Act of 1934 during the preceding 12 months (or for such shorter period that the registrant was required to file such reports), and (2) has been subject to such filing requirements for the past 90 days. Yes [X] No [ ]
Indicate by check mark whether the registrant has submitted electronically every Interactive Data File required to be submitted pursuant to Rule 405 of Regulation S-T during the preceding 12 months (or for such shorter period that the registrant was required to submit such files). Yes [X] No [ ]
Indicate by check mark whether the registrant is a large accelerated filer, an accelerated filer, a non-accelerated filer, a smaller reporting company, or emerging growth company. See definition of “large accelerated filer,” “accelerated filer,” “smaller reporting company,” and “emerging growth company” in Rule 12b-2 of the Exchange Act.
| Large accelerated filer | [ ] | Accelerated filer | [ ] |
| Non-accelerated filer | [ ] | Smaller reporting company | [X] |
| Emerging growth company | [ ] |
If an emerging growth company, indicate by check mark if the registrant has elected not to use the extended transition period for complying with any new or revised financial accounting standards provided pursuant to Section 13(a) of the Exchange Act. [ ]
Indicate by check mark whether the registrant is a shell company (as defined in Rule 12b-2 of the Exchange Act). Yes [ ] No [X]
The number of outstanding shares of the registrant’s common stock as of April 30, 2021 was 15,521,616.
PROCESSA PHARMACEUTICALS, INC.
TABLE OF CONTENTS
| 2 |
Processa Pharmaceuticals, Inc.
Condensed Consolidated Balance Sheets
(Unaudited)
| March 31, 2021 | December 31, 2020 | |||||||
| ASSETS | ||||||||
| Current Assets | ||||||||
| Cash and cash equivalents | $ | 23,048,245 | $ | 15,416,224 | ||||
| Due from related party | 3,566 | 154,730 | ||||||
| Due from tax agencies | 70,274 | 77,024 | ||||||
| Prepaid expenses and other | 1,424,138 | 554,708 | ||||||
| Total Current Assets | 24,546,223 | 16,202,686 | ||||||
| Property and Equipment, net | - | 484 | ||||||
| Other Assets | ||||||||
| Operating lease right-of-use assets, net of accumulated amortization | 138,108 | 158,558 | ||||||
| Intangible assets, net of accumulated amortization | 8,648,294 | 8,847,126 | ||||||
| Security deposit | 5,535 | 5,535 | ||||||
| Total Other Assets | 8,791,937 | 9,011,219 | ||||||
| Total Assets | $ | 33,338,160 | $ | 25,214,389 | ||||
| LIABILITIES AND STOCKHOLDERS’ EQUITY | ||||||||
| Current Liabilities | ||||||||
| Note payable – Paycheck Protection Program, current portion | $ | 144,888 | $ | 117,574 | ||||
| Current maturities of operating lease liability | 97,270 | 87,200 | ||||||
| Accrued interest | 1,312 | 950 | ||||||
| Accounts payable | 177,021 | 320,694 | ||||||
| Due to licensor | 400,000 | 400,000 | ||||||
| Due to related parties | 41,258 | 69,858 | ||||||
| Accrued expenses | 233,895 | 224,676 | ||||||
| Total Current Liabilities | 1,095,644 | 1,220,952 | ||||||
| Non-current Liabilities | ||||||||
| Note payable – Paycheck Protection Program | 17,571 | 44,885 | ||||||
| Non-current operating lease liability | 47,444 | 78,463 | ||||||
| Non-current due to licensor | 400,000 | 400,000 | ||||||
| Net deferred tax liability | 441,363 | 530,611 | ||||||
| Total Liabilities | 2,002,022 | 2,274,911 | ||||||
| Commitments and Contingencies | - | - | ||||||
| Stockholders’ Equity | ||||||||
| Common stock, par value $0.0001, 30,000,000 shares authorized: 15,521,616 and 14,187,984 issued and outstanding at March 31, 2021 and December 31, 2020, respectively | 1,552 | 1,419 | ||||||
| Additional paid-in capital | 58,829,865 | 48,333,857 | ||||||
| Accumulated deficit | (27,495,279 | ) | (25,395,798 | ) | ||||
| Total Stockholders’ Equity | 31,336,138 | 22,939,478 | ||||||
| Total Liabilities and Stockholders’ Equity | $ | 33,338,160 | $ | 25,214,389 | ||||
The accompanying notes are an integral part of these condensed consolidated financial statements.
| 3 |
Processa Pharmaceuticals, Inc.
Condensed Consolidated Statements of Operations
Three Months Ended March 31, 2021 and 2020
(Unaudited)
| Three months ended March 31, | ||||||||
| 2021 | 2020 | |||||||
| Operating Expenses | ||||||||
| Research and development expenses | $ | 1,476,160 | $ | 501,771 | ||||
| General and administrative expenses | 717,147 | 484,353 | ||||||
| Operating Loss | (2,193,307 | ) | (986,124 | ) | ||||
| Other Income (Expense) | ||||||||
| Interest expense | (362 | ) | (17,170 | ) | ||||
| Interest income | 4,940 | 829 | ||||||
| Net Operating Loss Before Income Tax Benefit | (2,188,729 | ) | (1,002,465 | ) | ||||
| Income Tax Benefit | 89,248 | 128,129 | ||||||
| Net Loss | $ | (2,099,481 | ) | $ | (874,336 | ) | ||
| Net Loss Per Common Share - Basic and Diluted | $ | (0.14 | ) | $ | (0.16 | ) | ||
| Weighted Average Common Shares Used to Compute Net Loss Per Common Shares - Basic and Diluted | 14,583,698 | 5,515,566 | ||||||
The accompanying notes are an integral part of these condensed consolidated financial statements.
| 4 |
Processa Pharmaceuticals, Inc.
Condensed Consolidated Statement of Changes in Stockholders’ Equity
Three Months Ended March 31, 2021 and 2020
(Unaudited)
| Additional | ||||||||||||||||||||
| Common Stock | Paid-In | Accumulated | ||||||||||||||||||
| Shares | Amount | Capital | Deficit | Total | ||||||||||||||||
| Balance at January 1, 2021 | 14,187,984 | $ | 1,419 | $ | 48,333,857 | $ | (25,395,798 | ) | $ | 22,939,478 | ||||||||||
| Stock-based compensation | 12,500 | 1 | 308,297 | - | 308,298 | |||||||||||||||
| Equity issued to a consultant as a prepayment for services | 312,293 | - | 312,293 | |||||||||||||||||
| Shares issued in private placement, net of transaction costs | 1,321,132 | 132 | 9,875,418 | - | 9,875,550 | |||||||||||||||
| Net loss | - | - | - | (2,099,481 | ) | (2,099,481 | ) | |||||||||||||
| Balance, March 31, 2021 | 15,521,616 | $ | 1,552 | $ | 58,829,865 | $ | (27,495,279 | ) | $ | 31,336,138 | ||||||||||
| Additional | Common Stock | |||||||||||||||||||||||
| Common Stock | Paid-In | Dividend | Accumulated | |||||||||||||||||||||
| Shares | Amount | Capital | Payable | Deficit | Total | |||||||||||||||||||
| Balance at January 1, 2020 | 5,486,595 | $ | 549 | $ | 18,994,008 | $ | 3 | $ | (10,982,010 | ) | $ | 8,012,550 | ||||||||||||
| Stock-based compensation | - | - | 98,663 | - | - | 98,663 | ||||||||||||||||||
| Transaction costs related to the 2020 underwritten public offering | - | - | (2,806 | ) | - | - | (2,806 | ) | ||||||||||||||||
| Net loss | - | - | - | - | (874,336 | ) | (874,336 | ) | ||||||||||||||||
| Balance, March 31, 2020 | 5,486,595 | $ | 549 | $ | 19,089,865 | $ | 3 | $ | (11,856,346 | ) | $ | 7,234,071 | ||||||||||||
The accompanying notes are an integral part of these condensed consolidated financial statements.
| 5 |
Processa Pharmaceuticals, Inc.
Condensed Consolidated Statements of Cash Flows
Three Months Ended March 31, 2021 and 2020
(Unaudited)
| 2021 | 2020 | |||||||
| Cash Flows From Operating Activities | ||||||||
| Net loss | $ | (2,099,481 | ) | $ | (874,336 | ) | ||
| Adjustments to reconcile net loss to net cash used in operating activities: | ||||||||
| Depreciation | 484 | 2,112 | ||||||
| Non-cash lease expense for right-of-use assets | 20,450 | 19,548 | ||||||
| Amortization of debt issuance costs | - | 1,070 | ||||||
| Amortization of intangible asset | 198,832 | 198,832 | ||||||
| Deferred income tax benefit | (89,248 | ) | (128,129 | ) | ||||
| Stock-based compensation | 308,298 | 98,663 | ||||||
| Net changes in operating assets and liabilities: | ||||||||
| Prepaid expenses and other | (557,137 | ) | 129,353 | |||||
| Operating lease liability | (20,949 | ) | (19,217 | ) | ||||
| Accrued interest | 362 | 16,100 | ||||||
| Accounts payable | (143,673 | ) | 17,604 | |||||
| Due (from) to related parties | 122,564 | (28,312 | ) | |||||
| Other receivables | 6,750 | - | ||||||
| Accrued expenses | 9,219 | 20,259 | ||||||
| Net cash used in operating activities | (2,243,529 | ) | (546,453 | ) | ||||
| Cash Flows From Financing Activities | ||||||||
| Net proceeds from private placement | 9,875,550 | - | ||||||
| Transaction costs related to fundraising efforts | - | (2,806 | ) | |||||
| Net cash (used in) provided by financing activities | 9,875,550 | (2,806 | ||||||
| Net Increase (Decrease) in Cash | 7,632,021 | (549,259 | ) | |||||
| Cash and Cash Equivalents – Beginning of Period | 15,416,224 | 691,536 | ||||||
| Cash and Cash Equivalents – End of Period | $ | 23,048,245 | $ | 142,277 | ||||
The accompanying notes are an integral part of these condensed consolidated financial statements.
| 6 |
Processa Pharmaceuticals, Inc.
Notes to Condensed Consolidated Financial Statements
(Unaudited)
Note 1 – Organization and Summary of Significant Accounting Policies
Business Activities and Organization
We are a clinical-stage biopharmaceutical company focused on the development of drug products that are intended to provide treatment for patients who have a high unmet medical need condition that effects survival or the patient’s quality of life and have few or no treatment options. We currently have three drugs in various stages of clinical development. Our most advanced product candidate, PCS499, is an oral tablet that is a deuterated analog of one of the major metabolites of pentoxifylline (PTX or Trental®). We have completed a Phase 2A trial for PCS499 and will begin recruiting patients for a Phase 2B trial in the second quarter of 2021. In addition, we have in-licensed PCS6422 (eniluracil) from Elion Oncology and PCS12852 from Yuhan Corporation. PCS6422 will be orally administered with capecitabine in a Phase 1B dose-escalation study in patients with Advanced Refractory Gastrointestinal (GI) Tract Tumors, with recruitment beginning in the second quarter of 2021. PCS12852 has already been evaluated in clinical studies in South Korea. We are planning on submitting an IND application in the third quarter of 2021 based on guidance received from the FDA for PCS12852 in patients with gastroparesis. Our remaining drug asset whose active molecules have been shown to be clinically efficacious require more toxicology data in order to more efficiently develop the drug clinically.
Impact of COVID-19
The extent of the impact of the COVID-19 pandemic on our business, operations and development timelines and plans remains uncertain, and will depend on certain developments, including the duration of the outbreak and its impact on our development activities, planned clinical trial enrollment, future trial sites, CROs, third-party manufacturers, and other third parties with whom we do business, as well as its impact on regulatory authorities and our key scientific and management personnel. Although we modified our operations and practices in 2020 due to the COVID-19 pandemic and to comply with federal, state and local requirements, our business, operations and development timelines were not materially adversely affected. However, the extent to which the COVID-19 pandemic may affect our business, operations and development timelines and plans in the future, including the resulting impact on our expenditures and capital needs, remains uncertain.
Basis of Presentation
The accompanying unaudited condensed consolidated financial statements have been prepared in accordance with accounting principles generally accepted in the United States of America (“U.S. GAAP”) for interim financial information and with the instructions of the Securities and Exchange Commission (“SEC”) on Form 10-Q and Article 8 of Regulation S-X.
Accordingly, they do not include all the information and disclosures required by U.S. GAAP for complete financial statements. All material intercompany accounts and transactions have been eliminated in consolidation. In the opinion of management, the accompanying unaudited condensed consolidated financial statements include all adjustments necessary, which are of a normal and recurring nature, for the fair presentation of the Company’s financial position and of the results of operations and cash flows for the periods presented. These condensed consolidated financial statements should be read in conjunction with the audited financial statements and notes thereto included in our Annual Report on Form 10-K for the year ended December 31, 2020, as filed with the SEC. The results of operations for the interim periods shown in this report are not necessarily indicative of the results that may be expected for any other interim period or for the full year.
| 7 |
Liquidity
We have incurred losses since inception, devoting substantially all of our efforts toward research and development, and have an accumulated deficit of approximately $27.5 million at March 31, 2021. During the three months ended March 31, 2021, we generated a net loss of approximately $2.1 million and we expect to continue to generate operating losses and negative cash flow from operations for the foreseeable future. However, we believe our cash balance at March 31, 2021 is adequate to fund our budgeted operations through 2023. Our ability to execute our longer-term operating plans, including unplanned future clinical trials for our portfolio of drugs depend on our ability to obtain additional funding from the sale of equity and/or debt securities, a strategic transaction or other funding transactions. We plan to continue to actively pursue financing alternatives, but there can be no assurance that we will obtain the necessary funding in the future when necessary.
We had no revenue during the three months ended March 31, 2021 and do not have any revenue under contract or any immediate sales prospects. Our primary uses of cash are to fund our planned clinical trials, research and development expenditures and operating expenses. Cash used to fund operating expenses is impacted by the timing of when we incur and pay these expenses.
Use of Estimates
In preparing our condensed consolidated financial statements and related disclosures in conformity with GAAP and pursuant to the rules and regulations of the SEC, we make estimates and judgments that affect the amounts reported in the condensed consolidated financial statements and accompanying notes. Estimates are used for, but not limited to preclinical and clinical trial expenses, stock-based compensation, intangible assets, future milestone payments and income taxes. These estimates and assumptions are continuously evaluated and are based on management’s experience and knowledge of the relevant facts and circumstances. While we believe the estimates to be reasonable, actual results could differ materially from those estimates and could impact future results of operations and cash flows.
Intangible Assets
Intangible assets acquired individually or with a group of other assets from others (other than in a business combination) are recognized at cost, including transaction costs, and allocated to the individual assets acquired based on relative fair values and no goodwill is recognized. Cost is measured based on cash consideration paid. If consideration given is in the form of non-cash assets, liabilities incurred, or equity interests issued, measurement of cost is based on either the fair value of the consideration given or the fair value of the assets (or net assets) acquired, whichever is more clearly evident and more reliably measurable. Costs of internally developing, maintaining or restoring intangible assets that are not specifically identifiable, have indeterminate lives or are inherent in a continuing business are expensed as incurred.
Intangible assets purchased from others for use in research and development activities and that have alternative future uses (in research and development projects or otherwise) are capitalized in accordance with ASC Topic 350, Intangibles – Goodwill and Other. Those that have no alternative future uses (in research and development projects or otherwise) and therefore no separate economic value are considered research and development costs and are expensed as incurred. Amortization of intangibles used in research and development activities is a research and development cost.
Intangibles with a finite useful life are amortized using the straight-line method unless the pattern in which the economic benefits of the intangible assets are consumed or used up are reliably determinable. The useful life is the best estimate of the period over which the asset is expected to contribute directly or indirectly to our future cash flows. The useful life is based on the duration of the expected use of the asset by us and the legal, regulatory or contractual provisions that constrain the useful life and future cash flows of the asset, including regulatory acceptance and approval, obsolescence, demand, competition and other economic factors. We evaluate the remaining useful life of intangible assets each reporting period to determine whether any revision to the remaining useful life is required. If the remaining useful life is changed, the remaining carrying amount of the intangible asset will be amortized prospectively over the revised remaining useful life. If an income approach is used to measure the fair value of an intangible asset, we consider the period of expected cash flows used to measure the fair value of the intangible asset, adjusted as appropriate for company-specific factors discussed above, to determine the useful life for amortization purposes.
If no regulatory, contractual, competitive, economic or other factors limit the useful life of the intangible to us, the useful life is considered indefinite. Intangibles with an indefinite useful life are not amortized until its useful life is determined to be no longer indefinite. If the useful life is determined to be finite, the intangible is tested for impairment and the carrying amount is amortized over the remaining useful life in accordance with intangibles subject to amortization. Indefinite-lived intangibles are tested for impairment annually and more frequently if events or circumstances indicate that it is more-likely-than-not that the asset is impaired.
| 8 |
Impairment of Long-Lived Assets and Intangibles Other Than Goodwill
We account for the impairment of long-lived assets in accordance with ASC 360, Property, Plant and Equipment and ASC 350, Intangibles – Goodwill and Other, which require that long-lived assets and certain identifiable intangibles be reviewed for impairment whenever events or changes in circumstances indicate that the carrying amount of an asset may not be recoverable. Recoverability of assets to be held and used is measured by a comparison of the carrying amount of an asset to its expected future undiscounted net cash flows generated by the asset. If such assets are considered to be impaired, the impairment to be recognized is measured as the amount by which the carrying amounts of the assets exceed the fair value of the assets based on the present value of the expected future cash flows associated with the use of the asset. Assets to be disposed of are reported at the lower of the carrying amount or fair value less costs to sell. Based on management’s evaluation, there was no impairment loss recorded during the three months ended March 31, 2021 or 2020.
Stock-based Compensation
Stock-based compensation expense is based on the grant-date fair value estimated in accordance with the provisions of ASC 718, Compensation-Stock Compensation. We expense stock-based compensation over the requisite service period based on the estimated grant-date fair value of the awards. Stock-based awards with graded-vesting schedules are recognized on a straight-line basis over the requisite service period for each separately vesting portion of the award. We estimate the fair value of stock option and warrant grants using the Black-Scholes option pricing model, and the assumptions used in calculating the fair value of stock-based awards represent management’s best estimates and involve inherent uncertainties and the application of management’s judgment. Stock-based compensation costs are recorded as general and administrative or research and development costs in the statements of operations based upon the underlying individual’s or consultant’s role.
Net Loss Per Share
Basic loss per share is computed by dividing our net loss available to common shareholders by the weighted average number of shares of common stock outstanding during the period. Diluted loss per share is computed by dividing our net loss available to common shareholders by the diluted weighted average number of shares of common stock during the period. Since we experienced a net loss for both periods presented, basic and diluted net loss per share are the same. As such, diluted loss per share for the three months ended March 31, 2021 and 2020 excludes the impact of potentially dilutive common shares related to outstanding stock options and warrants and, in 2020, the conversion of our 2019 Senior Notes since those shares would have an anti-dilutive effect on loss per share.
In December 2019, we determined the sale of the 2019 Senior Notes triggered the full ratchet anti-dilution provision of the common stock we sold in 2018 Private Placement Transactions. As a result, those shareholders were entitled to 28,971 shares of common stock in the fourth quarter of 2019. At March 31, 2020, we accounted for these shares as a common stock dividend payable as they were not issued until June 2020. For purposes of computing our basic and diluted EPS, we included the shares in our weighted number of common shares outstanding for the three months ended March 31, 2020.
Our diluted net loss per share for the three months ended March 31, 2021 and 2020 excluded 989,531 and 762,157 of potentially dilutive common shares, respectively, related to outstanding stock options, warrants and unvested restricted stock and, in 2020, the conversion of our Senior Notes since those shares would have had an anti-dilutive effect on loss per share during the periods then ended.
Recent Accounting Pronouncements
From time to time, the Financial Accounting Standards Board (“FASB”) or other standard setting bodies issue new accounting pronouncements. Updates to the FASB Accounting Standards Codification are communicated through issuance of an Accounting Standards Update (“ASU”). We have implemented all new accounting pronouncements that are in effect and that may impact our condensed consolidated financial statements. We have evaluated recently issued accounting pronouncements and determined that there is no material impact on our financial position or results of operations.
| 9 |
Note 2 – Property and Equipment
Property and equipment at March 31, 2021 and December 31, 2020 consisted of the following:
| March 31, 2021 | December 31, 2020 | |||||||
| Software | 19,740 | 19,740 | ||||||
| Office equipment | 9,327 | 9,327 | ||||||
| Total Cost | 29,067 | 29,067 | ||||||
| Less: accumulated depreciation | 29,067 | 28,583 | ||||||
| Property and equipment, net | - | 484 | ||||||
Note 3 – Intangible Assets
Intangible assets at March 31, 2021 and December 31, 2020 consisted of the following:
| March 31, 2021 | December 31, 2020 | |||||||
| Gross intangible assets | $ | 11,059,429 | $ | 11,059,429 | ||||
| Less: accumulated amortization | (2,411,135 | ) | (2,212,303 | ) | ||||
| Total intangible assets, net | $ | 8,648,294 | $ | 8,847,126 | ||||
Amortization expense was $198,832 for the three months ended March 31, 2021 and 2020 and is included within research and development expense in the accompanying condensed consolidated statements of operations. As of March 31, 2021, our estimated amortization expense for the 2021 will be approximately $790,000 and approximately $788,000 per year for annual periods thereafter.
The capitalized costs for the license rights to PCS499 included the $8 million purchase price, $1,782 in transaction costs and $3,037,147 associated with the initial recognition of an offsetting deferred tax liability related to the acquired temporary difference for an asset purchased that is not a business combination and has a tax basis of $1,782 in accordance with ASC 740-10-25-51 Income Taxes. In accordance with ASC Topic 730, Research and Development, we capitalized the costs of acquiring the exclusive license rights to PCS499, as the exclusive license rights represent intangible assets to be used in research and development activities that management believes has future alternative uses.
Note 4 – Income Taxes
We account for income taxes in accordance with ASC Topic 740, Income Taxes. Deferred income taxes are recorded for the expected tax consequences of temporary differences between the tax basis of assets and liabilities for financial reporting purposes and amounts recognized for income tax purposes. As of March 31, 2021 and December 31, 2020, we recorded a valuation allowance equal to the full recorded amount of our net deferred tax assets related to deferred start-up costs, federal orphan drug tax credit and certain other minor temporary differences since it is more-likely-than-not that such benefits will not be realized. The valuation allowance is reviewed quarterly and is maintained until sufficient positive evidence exists to support its reversal.
A deferred tax liability was recorded on March 19, 2018 when Processa received CoNCERT’s license and “Know-How” in exchange for Processa stock that had been issued in the Internal Revenue Code Section 351 Transaction. The Section 351 Transaction treats the acquisition of the license and Know-How for stock as a tax-free exchange. As a result, under ASC 740-10-25-51 Income Taxes, Processa recorded a deferred tax liability of $3,037,147 for the acquired temporary difference between intangible assets (see Note 3) for the financial reporting basis of $11,038,929 and the tax basis of $1,782. The deferred tax liability will be reduced for the effect of non-deductibility of the amortization of the intangible asset and may be offset by the deferred tax assets resulting from net operating tax losses.
| 10 |
Under ACS 740-270 Income Taxes – Interim Reporting, we are required to project our annual federal and state effective income tax rate and apply it to the year-to-date ordinary operating tax basis loss before income taxes. Based on the projection, we expect to recognize the tax benefit from our projected ordinary tax loss, which can be used to offset the deferred tax liabilities related to the intangible assets and resulted in the recognition of a deferred tax benefit shown in the condensed consolidated statements of operations for three months ended March 31, 2021 and 2020. No current income tax expense is expected for the foreseeable future as we expect to generate taxable net operating losses.
Note 5 – Stock-based Compensation
We did not grant any stock options to employees or non-employees during the three months ended March 31, 2021 or 2020. At March 31, 2021, we had outstanding options to purchase 152,806 shares of our common stock of which options for the purchase of 115,750 shares of our common stock were vested. All the options outstanding have exercise prices higher than the closing market price at March 31, 2021.
At March 31, 2021, we also had 122,782 shares of unvested restricted stock that was granted in 2020 outstanding.
During the three months ended March 31, 2021, we issued 12,500 restricted stock units (RSUs) and 100,000 warrants to a consultant for services to be provided in 2021. We valued the RSUs based on the closing share price on the date of grant. The fair value of the warrants granted was estimated using the Black-Scholes option pricing model at the date of grant. We also agreed to grant 64,556 RSUs and 30,000 stock options to our employees and a consultant in accordance with our employment/consulting agreements. These RSUs and stock options are subject to shareholder approval; thus, they are not included in the number of potentially dilutive securities.
We recorded $308,298 and $98,663 of stock-based compensation expense for the three months ended March 31, 2021 and 2020, respectively.
Note 6 – Paycheck Protection Program Loan
In May 2020, we entered into a $162,459 Paycheck Protection Promissory Note (the “PPP Loan”) with the Bank of America. The PPP Loan was made under, and is subject to the terms and conditions of, the PPP which was established under the CARES Act and is administered by the U.S. Small Business Administration. The current term of the loan is two years with a maturity date of May 5, 2022 and it contains a favorable fixed annual interest rate of 1.00%. Payments of principal and interest on the PPP Loan were deferred for the first six months of the term of the PPP Loan until November 5, 2020. Principal and interest are payable monthly and may be prepaid by us at any time prior to maturity with no prepayment penalties. Under the terms of the CARES Act, recipients can apply for and receive forgiveness for all, or a portion, of the loan granted under the PPP. Such forgiveness will be determined, subject to limitations, based on the use of loan proceeds for certain permissible purposes as set forth in the PPP, including, but not limited to, payroll costs, mortgage interest, rent or utility costs, and on the maintenance of employee and compensation levels during a certain time period following the funding of the PPP Loan. We used the entire proceeds of our PPP Loan for payroll costs and applied for full forgiveness on January 18, 2021. On February 11, 2021, Bank of America submitted a decision to the Small Business Administration that the full amount should be forgiven. However, no assurance is provided that we will be able to obtain full or partial forgiveness of the PPP Loan. As of March 31, 2021, $144,888 of the total $162,459 PPP-related debt is classified as a current liability on our consolidated balances sheet.
Note 7 – Stockholders’ Equity
On January 11, 2021, we granted 12,500 RSUs and 100,000 warrants to a consultant for services to be provided in 2021. We recorded $312,293 as a prepaid expense for the portion of services the consultant has yet to provide.
On February 24, 2021, we sold in a private placement 1,321,132 shares of our common stock to accredited and institutional investors for gross proceeds of $10.2 million. Net proceeds from the offering were $9.9 million. We issued warrants for the purchase of 79,268 shares of our common stock to our placement agent. These warrants are exercisable for cash at $9.30 per share and expire on February 24, 2023.
We have not had any sales of our preferred stock since we were incorporated on March 29, 2011 and there were no issued or outstanding shares of preferred stock at March 31, 2021 or December 31, 2020.
Note 8 – Net Loss per Share of Common Stock
Basic net loss per share is computed by dividing net loss by the weighted average common shares outstanding. Diluted net loss per share is computed by dividing net loss by the weighted average common shares outstanding, which includes potentially dilutive effect of stock options, warrants and senior convertible notes. Since we experienced a loss for both periods presented, any dilutive common shares outstanding were excluded from the computation as shown below, as they would have an anti-dilutive impact on diluted net loss per share. The treasury-stock method is used to determine the dilutive effect of our stock options and warrants grants, and the if-converted method is used to determine the dilutive effect of the Senior Notes.
| 11 |
The computation of net loss per share for the three months ended March 31, 2021 and 2020 was as follows:
Three months ended March 31, | ||||||||
| 2021 | 2020 | |||||||
| Basic and diluted net loss per share: | ||||||||
| Net loss | $ | (2,099,481 | ) | $ | (874,336 | ) | ||
| Weighted average number of common shares-basic and diluted | 14,583,698 | 5,515,566 | ||||||
| Basic and diluted net loss per share | $ | (0.14 | ) | $ | (0.16 | ) | ||
As described in Note 1, in December 2019, we determined the sale of the 2019 Senior Notes triggered the full ratchet anti-dilution provision of the common stock we sold in 2018 Private Placement Transactions. As a result, those shareholders were entitled to 28,971 shares of common stock in the fourth quarter of 2019. At March 31, 2020, we accounted for these shares as a common stock dividend payable as they were not issued until June 2020. For purposes of computing our basic and diluted EPS, we included the shares in our weighted number of common shares outstanding for the three months ended March 31, 2020.
The following potentially dilutive securities were excluded from the computation of diluted net income per share as their effect would have been anti-dilutive for the periods presented.
| 2021 | 2020 | |||||||
| Stock options and purchase warrants | 989,531 | 704,657 | ||||||
| Senior convertible notes | - | 57,500 | ||||||
Note 9 – Operating Leases
We lease our office space under an operating lease agreement. This lease does not have significant rent escalation, concessions, leasehold improvement incentives, or other build-out clauses. Further, the lease does not contain contingent rent provisions. We also lease office equipment under an operating lease. Our office space lease includes both lease (e.g., fixed payments including rent, taxes, and insurance costs) and non-lease components (e.g., common-area or other maintenance costs), which are accounted for as a single lease component as we have elected the practical expedient to group lease and non-lease components for all leases. Our leases do not provide an implicit rate and, as such, we have used our incremental borrowing rate of 8% in determining the present value of the lease payments based on the information available at the lease commencement date.
Lease costs included in our condensed consolidated statements of operations totaled $24,030 and $24,207 for the three months ended March 31, 2021 and 2020, respectively. The weighted average remaining lease terms and discount rate for our operating leases were as follows at March 31, 2021:
| Weighted average remaining lease term (years) for our facility and equipment leases | 1.7 | |||
| Weighted average discount rate for our facility and equipment leases | 8.00 | % |
Maturities of our lease liabilities for all operating leases were as follows as of March 31, 2021:
| 2021 | $ | 72,750 | ||
| 2022 | 75,969 | |||
| 2023 | 6,228 | |||
| 2024 | 1,557 | |||
| Total lease payments | 156,504 | |||
| Less: Interest | (11,790 | ) | ||
| Present value of lease liabilities | 144,714 | |||
| Less: current maturities | (97,270 | ) | ||
| Non-current lease liability | $ | 47,444 |
Note 10 – Related Party Transactions
A shareholder, CorLyst, LLC, reimburses us for shared costs related to payroll, health care insurance and rent based on actual costs incurred, which are recognized as a reduction of our general and administrative operating expenses in our condensed consolidated statement of operations. In September 2020, CorLyst prepaid shared expenses to us for the fourth quarter of 2020 through the second quarter of 2021. At March 31, 2021, we recognized $41,258 in prepaid reimbursements as due to related parties in the accompanying condensed consolidated balance sheet. No amounts were due from CorLyst at March 31, 2021 and December 31, 2020.
Note 11 – Commitments and Contingencies
Purchase Obligations
We enter into contracts in the normal course of business with contract research organizations and subcontractors to further develop our products. The contracts are cancellable, with varying provisions regarding termination. If we terminated a cancellable contract with a specific vendor, we would only be obligated for products or services that we received as of the effective date of the termination and any applicable cancellation fees. We are contractually obligated to pay up to approximately $5.1 million of future services under the agreements with the CROs, but our actual contractual obligations will vary depending on the progress and results of the clinical trials.
| 12 |
Item 2. Management’s Discussion and Analysis of Financial Condition and Results of Operation
Forward Looking Statements
This Quarterly Report on Form 10-Q contains “forward-looking statements” that reflect, when made, the Company’s expectations or beliefs concerning future events that involve risks and uncertainties. Forward-looking statements frequently are identified by the words “believe,” “anticipate,” “expect,” “estimate,” “intend,” “project,” “will be,” “will continue,” “will likely result,” or other similar words and phrases. Similarly, statements herein that describe the Company’s objectives, plans or goals also are forward-looking statements. Actual results could differ materially from those projected, implied or anticipated by the Company’s forward-looking statements. Some of the factors that could cause actual results to differ include: our limited operating history, limited cash and history of losses; our ability to achieve profitability; our ability to obtain adequate financing to fund our business operations in the future; the impact of the global pandemic caused by the novel coronavirus, COVID-19, including its impact on our ability to obtain financing or complete clinical trials; our ability to secure required FDA or other governmental approvals for our product candidates and the breadth of the indication sought; the impact of competitive or alternative products, technologies and pricing; whether we are successful in developing and commercializing our technology, including through licensing; the adequacy of protections afforded to us and/or our licensor by the anticipated patents that we own or license and the cost to us of maintaining, enforcing and defending those patents; our and our licensor’s ability to protect non-patented intellectual property rights; our exposure to and ability to defend third-party claims and challenges to our and our licensor’s anticipated patents and other intellectual property rights; and our ability to continue as a going concern. For a discussion of these and all other known risks and uncertainties that could cause actual results to differ from those contained in the forward-looking statements, see “Risk Factors” in the Company’s Annual Report on Form 10-K for the year ended December 31, 2020, which is available on the SEC’s website at www.sec.gov. All forward-looking statements are qualified in their entirety by this cautionary statement, and the Company undertakes no obligation to revise or update this Quarterly Report on Form 10-Q to reflect events or circumstances after the date hereof.
For purposes of this Management’s Discussion and Analysis of Financial Condition and Results of Operations, references to the “Company,” “we,” “us” or “our” refer to the operations of Processa Pharmaceuticals, Inc. and its direct and indirect subsidiaries for the periods described herein.
Overview
We are a clinical-stage biopharmaceutical company focused on the development of drug products that are intended to provide treatment for patients who have a high unmet medical need condition that effects survival or the patient’s quality of life and have few or no treatment options. We currently have three drugs in various stages of clinical development. Our most advanced product candidate, PCS499, is an oral tablet that is a deuterated analog of one of the major metabolites of pentoxifylline (PTX or Trental®). We have completed a Phase 2A trial for PCS499 and will begin recruiting patients for a Phase 2B trial in the second quarter of 2021. In addition, we have in-licensed PCS6422 (eniluracil) from Elion Oncology and PCS12852 from Yuhan Corporation. PCS6422 will be orally administered with capecitabine in a Phase 1B dose-escalation study in patients with Advanced Refractory Gastrointestinal (GI) Tract Tumors, with recruitment beginning in the second quarter of 2021. PCS12852 has already been evaluated in clinical studies in South Korea. We are planning on submitting an IND application in the third quarter of 2021 based on guidance received from the FDA on the clinical development program for PCS12852 in patients with gastroparesis. Our remaining drug asset whose active molecules have been shown to be clinically efficacious require more toxicology data in order to more efficiently develop the drug clinically.
Our Strategy
Our strategy is to acquire or in-license development candidates that will not only treat a specific group of patients with unmet medical needs, but may also have the potential to chart a more efficient path to registration. In many instances, these clinical candidates have significant pre-clinical and clinical data that we can leverage to high value inflection points while de-risking the programs and adding in optionality to potential future indications. The regulatory science approach our team has developed seeks to leverage the earlier data and identify the least risk path toward commercialization/registration of these drugs. We apply rigorous standards to identify drugs for our portfolio, namely:
| i. | The drug must represent a treatment option to patients with a high unmet medical need condition by improving survival and/or quality of life for these patients, |
| 13 |
| ii. | The drug or its metabolite or a drug with similar pharmacological properties must have demonstrated some evidence of efficacy in the target population, and | |
| iii. | The drug can be quickly developed such that within 2-4 years, critical value-added clinical milestones can be achieved while advancing the drug closer to commercialization and adding to the potential for a high return on investment. |
In order to add significant value to our in-licensed drugs within 2 to 4 years, the drugs must be in the clinical development stage and not in discovery stage, and we must be able to obtain clinical data to support the added value during those 2 to 4 years. The additional clinical data could range from a clinical proof-of-concept data to further demonstrate that the proposed pharmacology occurs clinically in the targeted patient population to a pivotal well-designed randomized controlled trial.
Our portfolio specifically includes drugs that (i) already have clinical proof-of-concept data demonstrating the desired pharmacological activity in humans or, minimally, clinical evidence in the form of case studies or clinical experience demonstrating the drug or a similar drug pharmacologically can successfully treat patients with the targeted indication; (ii) target indications for which the FDA believes that a single positive pivotal study demonstrating efficacy provides enough evidence that the clinical benefits of the drug and its approval outweighs the risks associated with the drug or the present standard of care (e.g., some orphan indications, many serious life-threatening conditions, some serious quality of life conditions); and/or (iii) target indications where the prevalence of the condition and the likelihood of patients enrolling in a study meet the desired time-frame to demonstrate that the drug can, at some level, treat or potentially treat patients with the condition.
To advance our mission, we have assembled an experienced and talented management and product development team. Our team is experienced in developing drug products through all principal regulatory tiers from IND enabling studies to NDA submission. The combined scientific, development and regulatory experience of our team members has resulted in more than 30 drug approvals by the FDA, over 100 meetings with the FDA and involvement with more than 50 drug development programs, including drug products targeted to patients who have an unmet medical need. Although we believe that the skills and experience of our team members in drug development and commercialization is an important indicator of our future success, the past successes of our team members in developing and commercializing pharmaceutical products does not guarantee that they will successfully develop and commercialize drugs for us. In addition, the growth in revenues of companies at which our executive officers and directors served in was due to many factors and does not guarantee that they will successfully operate or manage us or that we will experience similar growth in revenues, even if they continue to serve as executive officers and/or directors.
Our ability to generate meaningful revenue from any products depends on our ability to out-license the drugs before or after we obtain FDA NDA approval. Even if our products are authorized and approved by the FDA, it should be noted that the products must still meet the challenges of successful marketing, distribution and consumer acceptance.
Impact of COVID-19
The COVID-19 pandemic has created uncertainties in the expected timelines for clinical stage biopharmaceutical companies such as ours, including possible delays in clinical trials and disruptions in the supply chain for raw materials used in clinical trial work. Such delays could materially impact our business in future periods. Furthermore, the spread of COVID-19, which has caused a broad impact globally, may materially affect us economically. While the potential economic impact brought by, and the duration of, COVID-19 may be difficult to assess or predict, a widespread pandemic could result in significant disruption of global financial markets, reducing our ability to access capital, which could in the future negatively affect our liquidity. Policymakers around the globe have responded with fiscal policy actions to support the healthcare industries and economies as a whole. The magnitude and overall effectiveness of these actions remain uncertain. Accordingly, the extent to which the COVID-19 global pandemic impacts our business, results of operations and financial condition will depend on future developments, which are highly uncertain and are difficult to predict. These developments include, but are not limited to, the duration and spread of the outbreak, its severity, the actions to contain the virus or address its impact, U.S. and foreign government actions to respond to the reduction in global economic activity, and how quickly and to what extent normal economic and operating conditions can resume. For more information on the risks associated with COVID-19, refer to Part I, Item 1A, “Risk Factors” in our Annual Report on Form 10-K.
| 14 |
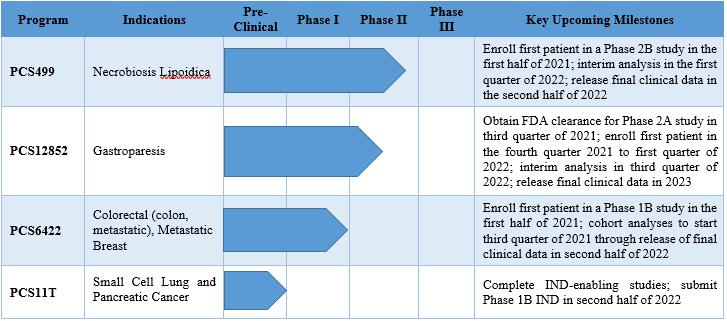
Our Drug Pipeline
The table below of our clinical product pipeline summarizes each drug, organized by phase of development. It should be noted that we expect the Phase 2B PCS499 and Phase 1B PCS6422 studies to have their first patient enrolled in the second quarter of 2021. The PCS12852 Phase 2A study will likely have first patient enrolled at the end of 2021 or beginning of 2022, depending on the submission and approval of our IND application.
PCS499
Our most advanced product candidate, PCS499, is an oral tablet that is a deuterated analog of one of the major metabolites of pentoxifylline (PTX or Trental®). PCS499 is classified by FDA as a new molecular entity. PCS499 and its metabolites act on multiple pharmacological targets that are important in a variety of conditions. We have targeted Necrobiosis Lipoidica (NL) as our lead indication for PCS499. NL is a chronic, disfiguring condition affecting the skin and tissue under the skin typically on the lower extremities with no currently approved FDA treatments. NL presents more commonly in women than in men and occurs more often in people with diabetes. Ulceration occurs in approximately 30% of NL patients, which can lead to more severe complications, such as deep tissue infections and osteonecrosis threatening the life of the limb. Approximately 22,000 - 55,000 people in the United States and more than 120,000 people outside the United States are affected with ulcerated NL.
The degeneration of tissue occurring at the NL lesion site may be caused by a number of pathophysiological changes, which make it extremely difficult to develop effective treatments for this condition. Because PCS499 and its metabolites appear to affect most of the biological pathways that contribute to the pathophysiology associated with NL, PCS499 may provide a novel treatment solution for NL.
On June 18, 2018, the FDA granted orphan-drug designation for PCS499 for the treatment of NL. On September 28, 2018, the IND for PCS499 in NL became effective, such that we initiated and completed a Phase 2A multicenter, open-label prospective trial designed to determine the safety and tolerability of PCS499 in patients with NL. The study initially had a six-month treatment phase and a six-month optional extension phase. In December 2019, we informed patients and sites that the study would conclude after the treatment phase and there would no longer be an extension phase. The first enrolled NL patient in this Phase 2A clinical trial was dosed on January 29, 2019 and the study completed enrollment on August 23, 2019. The last patient visit took place in February 2020. Due to COVID-19 related restrictions at certain sites, study closeout, database lock and final report were delayed and completed in December 2020.
| 15 |
The primary objective of the Phase 2A trial was to evaluate the safety and tolerability of PCS499 in patients with NL and to use the safety and efficacy data to design future clinical trials. Based on toxicology studies and healthy human volunteer studies, Processa and the FDA agreed that a PCS499 dose of 1.8 grams/day would be the highest dose administered to NL patients in this Phase 2A trial. As anticipated, the PCS499 dose of 1.8 grams/day, 50% greater than the maximum tolerated dose of PTX, appeared to be well tolerated with no serious adverse events (SAEs) reported. All adverse events (AEs) reported in the study were mild in severity. As expected, gastrointestinal symptoms were the most frequent adverse events and reported in four patients, all of which resolved within 1-2 weeks of starting dosing.
Two of the twelve patients in the study presented with more severe ulcerated NL and had ulcers for more than two months prior to dosing. At baseline, the reference ulcer in one of the two patients measured 3.5 cm2 and had completely closed by Month 2 of treatment. The second patient had a baseline reference ulcer of 1.2 cm2 which completely closed by Month 9 during the patient’s treatment extension period. In addition, while in the trial, both patients also developed small ulcers at other sites, possibly related to contact trauma, and these ulcers resolved within one month. The other ten patients, presenting with mild to moderate NL and no ulceration, had more limited improvement of the NL lesions during treatment. Historically, 13 - 20% of all the patients with NL naturally progress to complete healing over many years after presenting with NL. Although the natural healing of the more severe ulcerated NL patients has not been evaluated independently, medical experts who treat NL patients suggest that the natural progression of an open ulcerated wound to complete closure would be significantly less than 13% over 1-2 years and probably close to 0% in patients with the larger ulcers.
On March 25, 2020, we met with the FDA and discussed the clinical program, as well as the nonclinical and clinical pharmacology plans to ultimately support the submission of the PCS499 New Drug Application (NDA) in the U.S. for the treatment of ulcers in NL patients. With input from the FDA, we have designed the next trial as a randomized, placebo-controlled Phase 2B study to evaluate the ability of PCS499 to completely close ulcers in patients with NL and better understand the potential response of NL patients on drug and on placebo. We have selected several clinical trial sites in the United States and are evaluating the inclusion of additional clinical trial sites, both within and outside of the United States. We will begin recruiting for the randomized, placebo-controlled trial in the second quarter of 2021 and are planning an interim analysis of the data from this trial in the first quarter of 2022. After obtaining the results from this Phase 2B study, we expect to have an end of Phase 2 meeting with the FDA to agree on the design of the Phase 3 study, to define a Special Protocol Assessment for the Phase 3 study and to agree on the next steps to obtain approval.
PCS12852
On August 19, 2020, we in-licensed PCS12852 (formerly known as YH12852) from Yuhan Corporation (“Yuhan”), pursuant to which we acquired an exclusive license to develop, manufacture and commercialize PCS12852 globally, excluding South Korea.
PCS12852 is a novel, potent and highly selective 5-hydroxytryptamine 4 (5-HT4) receptor agonist. Other 5-HT receptor agonists with less 5-HT4 selectivity have been shown to successfully treat gastrointestinal (GI) motility disorders such as gastroparesis, chronic constipation, constipation-predominant irritable bowel syndrome, and functional dyspepsia. Less selective 5-HT4 agonists, such as cisapride, have been either removed from the market or not approved because of the cardiovascular side effects associated with the drugs binding to other receptors, especially receptors other than 5-HT4.
| 16 |
We plan to submit an IND application in the third quarter of 2021 based on guidance received from the FDA on the clinical development program required for patients with gastroparesis. We plan to conduct a Phase 2A randomized, placebo-controlled study in patients with gastroparesis. The purpose of the Phase 2A trial is to evaluate the safety, efficacy and pharmacokinetics of two different dosing regimens for PCS12852. Data obtained from this study will be used to better design a future Phase 2/3 efficacy study. Since patients with gastroparesis have an abnormal pattern of upper GI motility in the absence of mechanical obstruction, the Phase 2A study will be designed to evaluate the change on gastric emptying in patients with gastroparesis on the two different dosing regimens of PCS12852 compared to placebo. The only FDA-approved drug to treat gastroparesis is metoclopramide, a dopamine D2 receptor antagonist that has serious side effects and can only be used as a short-term treatment. Other 5-HT4 drugs have been used clinically but the side effects, caused mainly by binding to other receptors, has resulted in these drugs not being a viable option to treat patients with gastroparesis. It should be noted that PCS12852 is a highly specific 5-HT4 agonist that has been shown in non-clinical studies to have a cardiovascular side effect only at concentrations greater than 1,000 times the maximum concentration seen in humans.
Two clinical studies, both which have demonstrated the effectiveness of PCS12852 on GI motility, have been previously conducted by Yuhan with PCS12852. In a Phase 1 trial (Protocol YH12852-101), the initial safety and tolerability of PCS12852 were evaluated after single and multiple oral doses in healthy subjects. PCS12852 was shown to increase GI motility in this study, increasing stool frequency with faster onset when compared to prucalopride, a less specific 5-HT4 agonist FDA-approved drug for the treatment of chronic idiopathic constipation. Based on an increase of ≥1 spontaneous bowel movement (SBM)/week from baseline during 7-day multiple dosing, the PCS12852 dose group had a higher percent of patients with an increase than the prucalopride group. All doses of PCS12852 were safe and well tolerated and no SAEs occurred during the study. The most frequently reported AEs were headache, nausea and diarrhea which were temporal, manageable, and reversible within 24 hours. There were no clinically significant changes in platelet aggregation and ECG parameters including a change in QTc prolongation in the study. In a Phase 1/2A clinical trial (Protocol YH12852-102), the safety, tolerability, gastric emptying rate and pharmacokinetics of multiple doses of a PCS12852 immediate release (IR) formulation and a delayed release (DR) formulation were evaluated. PCS12852 was safe and well tolerated after single and multiple administrations. The most frequent AEs for both the IR and DR formulations of PCS12852 were headache, nausea and diarrhea, but the incidences of these AEs were comparable with those of the 2mg prucalopride group. These AEs, which were transient and mostly mild in severity, are also commonly observed with other 5-HT4 agonists. Both formulations of PCS12852 also increased the gastric emptying rate and increased GI motility.
Yuhan had also conducted extensive toxicological studies for the product that demonstrated that the product is safe for use and can be moved into Phase 2 studies.
PCS6422
On August 23, 2020, we in-licensed PCS6422 from Elion Oncology, Inc. (“Elion”), pursuant to which we acquired an exclusive license to develop, manufacture and commercialize PCS6422 globally.
Elion acquired eniluracil (PCS6422) from Fennec Pharmaceuticals (formerly known as Adherex Technologies) in 2016. PCS6422 is an oral, potent, selective and irreversible inhibitor of dihydropyrimidine dehydrogenase (DPD), the enzyme that rapidly metabolizes a common chemotherapy drug known as 5-FU, into inactive metabolites, such as α-fluoro-β-alanine (F-Bal). F-Bal is a metabolite that has no anti-cancer activity but causes unwanted side effects, which notably leads to dose interruptions and significantly affect a patient’s quality of life. F-Bal is thought to cause the neurotoxicity and Hand–Foot Syndrome (HFS) associated with 5-FU, and greater formation of F-Bal appears to be associated with a decrease in the antitumor activity of 5-FU. HFS can affect activities of daily living, quality of life, and requires dose interruptions/adjustments and even therapy discontinuation resulting in suboptimal tumor effects. We believe that the inhibition of DPD by PCS6422 will significantly reduce 5-FU side effects related to F-Bal. One dose of PCS6422 irreversibly blocks DPD activity for up to two weeks until DPD levels recover via de novo synthesis. Thus, we believe inhibition of DPD will result in an improved safety profile given the decrease in F-Bal and potentially higher 5-FU intra-tumoral anti-cancer metabolites that could improve efficacy.
Fluoropyrimidines (e.g., 5-FU) remain the cornerstone of treatment for many different types of cancers, either as monotherapy or in combination with other chemotherapy agents by an estimated two million patients annually. Xeloda®, the brand name of capecitabane, is an oral pro-drug of 5-FU and approved as first-line therapy for metastatic colorectal and breast cancer. However, its use is limited by adverse effects such as the development of HFS in up to 60% of patients.
| 17 |
Elion evaluated the potential for the combination of PCS6422 with capecitabine as a treatment of advanced gastrointestinal (GI) tumors. Nonclinical efficacy data indicated that in colorectal cancer models, pretreatment with PCS6422 enhanced the antitumor activity of capecitabine. PCS6422 dramatically increased the antitumor potency of capecitabine without increasing the toxicity. The antitumor efficacy of the combination of PCS6422 and capecitabine was tested in several xenograft animal models with human breast, pancreatic and colorectal cancer cells. These preclinical xenograft models demonstrate that PCS6422 potentiates the antitumor activity of capecitabine and significantly reduces the dose of capecitabine required to be efficacious.
Elion met with the FDA in 2019 and agreed upon the clinical development program required for the combination of PCS6422 and capecitabine as first-line therapy for metastatic colorectal cancer when treatment with fluoropyrimidine therapy alone is preferred. On May 17, 2020, an IND for the Phase 1B study was granted safe to proceed by the FDA. This Phase 1B study will evaluate i) the safety and tolerability of escalating doses of capecitabine with a fixed dose of PCS6422 in advanced GI tumor patients, ii) the pharmacokinetics of PCS6422, capecitabine, 5-FU and selected metabolites, and iii) the activity of DPD over time after PCS6422 administration. We have selected several clinical trial sites in the United States for the study, which will begin patient recruitment in the second quarter of 2021.
Other DPD enzyme inhibitors (e.g. Gimeracil used in Teysuno® approved only outside the US) act as competitive reversible inhibitors. These agents must be present when 5-FU or capecitabine are administered to inhibit 5-FU breakdown by DPD in order to improve the efficacy and safety profiles of 5-FU. Given the reversible nature of their effect on DPD, over time 5-FU metabolism to F-Bal will return if the reversible inhibitor is not present, decreasing the amount of 5-FU in the cancer cells and decreasing the potential cytotoxicity on the cancer cells. There is also evidence that administering DPD inhibitors directly with 5-FU may also decrease the antitumor effect of the 5-FU. Because PCS6422 is an irreversible inactivator of DPD, it can be dosed the day before capecitabine administration and its effect on DPD can last longer than the reversible DPD inhibitors and beyond the time 5-FU exists in the cancer cell, even after PCS6422 has been completely eliminated out of the body. We believe this can optimize the potential cytotoxic effect and minimize the catabolism of 5-FU to F-Bal.
Prior to Elion’s involvement, two multicenter Phase 3 studies were conducted in patients with colorectal cancer with PCS6422 administered in 10-fold excess to 5-FU and administered with the 5-FU. Unfortunately, we believe the dose of PCS6422 during these trials was not optimal and that PCS6422 was not administered early enough to irreversibly affect the DPD enzyme, thus the regimen tended to produce less antitumor benefit than the control arm with the standard regimen of 5-FU/leucovorin (LV) without PCS6422. Later preclinical work suggested that when PCS6422 was present at the same time as and in excess to 5-FU, it diminished the antitumor activity of 5-FU, which we believe supports the proposal of exploring clinically dosing PCS6422 several hours before 5-FU to allow its complete clearance before the administration of 5-FU.
PCS11T
On May 24, 2020, we in-licensed PCS11T (formerly known as ATT-11T) from Aposense, Ltd. (“Aposense”), pursuant to which we were granted Aposense’s patent rights and Know-How to develop and commercialize their next generation irinotecan cancer drug, PCS11T.
PCS11T is a novel lipophilic anti-cancer pro-drug that is being developed for the treatment of the same solid tumors as prescribed for irinotecan. This pro-drug is a conjugate of a specific proprietary Aposense molecule connected to SN-38, the active metabolite of irinotecan. The proprietary molecule in PCS11T has been designed to allow PCS11T to bind to cell membranes to form an inactive pro-drug depot on the cell with SN-38 preferentially accumulating in the membrane of tumors cells and the tumor core. This unique characteristic may make the therapeutic window of PCS11T wider than other irinotecan products such that the antitumor effect of PCS11T could occur at a much lower dose with a milder adverse effect profile than irinotecan. Despite the widespread use of commercially marketed irinotecan products in the treatment of metastatic colorectal cancer and other cancers resulting in peak annual sales of approximately $1.1 billion, irinotecan has a narrow therapeutic window and includes an FDA “Black Box” warning for both neutropenia and severe diarrhea. There is, therefore, a substantial unmet need to overcome the limitations of the current commercially marketed irinotecan products, improving efficacy and reducing the severity of treatment emergent AEs. We believe the potential wider therapeutic window of PCS11T will likely lead to more patients responding with less side effects when on PCS11T compared to other irinotecan products.
| 18 |
Pre-clinical studies conducted to date showed that PCS11T demonstrated tumor eradication at much lower doses than irinotecan across various tumor xenograft models. PCS11T does not affect acetyl choline esterase (AChE) activity in human and rat plasma in vitro, which would suggest that PCS11T will show an improved safety profile, compared to irinotecan, which is known for its cholinergic-related side effects.
We are currently planning to manufacture the product at a GMP facility, conduct the required toxicological studies required to file the IND and initiate the Phase 1B study in oncology patients with solid tumors in 2022.
Results of Operations
Comparison of the three months ended March 31, 2021 and 2020
The following table summarizes our net loss during the periods indicated:
| Three months ended | ||||||||||||
| March 31, | ||||||||||||
| 2021 | 2020 | Change | ||||||||||
| Operating Expenses | ||||||||||||
| Research and development expenses | $ | 1,476,160 | $ | 501,771 | $ | 974,389 | ||||||
| General and administrative expenses | 717,147 | 484,353 | 232,794 | |||||||||
| Operating Loss | (2,193,307 | ) | (986,124 | ) | ||||||||
| Other Income (Expense) | ||||||||||||
| Interest expense | (362 | ) | (17,170 | ) | 16,808 | |||||||
| Interest income | 4,940 | 829 | 4,111 | |||||||||
| Net Operating Loss Before Income Tax Benefit | (2,188,729 | ) | (1,002,465 | ) | ||||||||
| Income Tax Benefit | 89,248 | 128,129 | (38,881 | ) | ||||||||
| Net Loss | $ | (2,099,481 | ) | $ | (874,336 | ) | ||||||
Revenues.
We do not currently have any revenue under contract or any immediate sales prospects.
Research and Development Expenses.
Our research and development costs are expensed as incurred. Research and development expenses include (i) licensing of compounds for product testing and development, (ii) program and testing related expenses, (iii) amortization of the exclusive license intangible asset used in research and development activities, and (iv) internal research and development staff related payroll, taxes and employee benefits, external consulting and professional fees related to the product testing and our development activities. Non-refundable advance payments for goods and services to be used in future research and development activities are recorded as prepaid expenses and expensed when the research and development activities are performed.
During the three months ended March 31, 2021, our research and development expenses increased by $974,389 to $1,476,160 from $501,771 when compared to the same period in 2020. Costs for the three months ended March 31, 2021 and 2020 were as follows:
| Three months ended March 31, | ||||||||
| 2021 | 2020 | |||||||
| Amortization of intangible assets | $ | 198,832 | $ | 198,832 | ||||
| Research and development salaries and benefits | 253,382 | 140,298 | ||||||
| Preclinical, clinical trial and other costs | 1,023,946 | 162,641 | ||||||
| Total | $ | 1,476,160 | $ | 501,771 | ||||
| 19 |
The increase in research and development expenses was primarily due to an increase of $861,305 in preclinical, clinical trial and other costs during the three months ended March 31, 2021 when compared to the same period in 2020. This increase was attributable to expenses we incurred as we commenced our Phase 2B clinical trial for PCS499, Phase 1B clinical trial for PCS6422 and for pre-IND costs for PCS12852. Expenses include costs we paid contract research organizations, for regulatory filing and maintenance fees, drug product testing and stability, consulting, and other clinical fees. During the same period in 2020, we were completing the patient portion of our Phase 2A clinical trial for PCS499 and incurring regulatory filing and consulting fees as we prepared for our meeting with the FDA. During the three months ended March 31, 2021, we also had increases in payroll and related costs of $113,084 from increased employee salary rates, hiring additional personnel and stock-based compensation, when compared to the same period in 2020.
We anticipate our research and development costs to increase significantly in the future as we start clinical trials for PCS499, PCS6422 and PCS12852, including the cost of having drug product manufactured, and the continued evaluation of the remaining drug in our portfolio.
The funding necessary to bring a drug candidate to market is subject to numerous uncertainties. Once a drug candidate is identified, the further development of that drug candidate may be halted or abandoned at any time due to a number of factors. These factors include, but are not limited to, funding constraints, safety or a change in market demand. For each of our drug candidate programs, we periodically assess the scientific progress and merits of the programs to determine if continued research and development is economically viable. Some programs may be terminated due to the lack of scientific progress and lack of prospects for ultimate commercialization. As noted above, we anticipate our research and development costs to increase in the future as we conduct the Phase 2B trial to evaluate the ability of PCS499 to completely close ulcers in patients with NL and begin a Phase 1B clinical trial for PCS6422. We expect to begin recruiting patients for these two clinical trials during the second quarter of 2021.
Our clinical trial cost accruals are based on estimates of patient enrollment and related costs at clinical investigator sites, as well as estimates for the services received and efforts expended pursuant to contracts with multiple research institutions and CROs that conduct and manage clinical trials on our behalf.
We estimate preclinical and clinical trial expenses based on the services performed, pursuant to contracts with research institutions and clinical research organizations that conduct and manage preclinical studies and clinical trials on our behalf. In accruing service fees, we estimate the time-period over which services will be performed and the level of patient enrollment and activity expended in each period. If the actual timing of the performance of services or the level of effort varies from the estimate, we will adjust the accrual accordingly. Payments made to third parties under these arrangements in advance of the receipt of the related series are recorded as prepaid expenses until the services are rendered.
General and Administrative Expenses.
Our general and administrative expenses for the three months ended March 31, 2021 increased by $232,794 to $717,147 from $484,353 for the three months ended March 31, 2020. The majority of the increase was an increase of $199,956 for professional fees, mostly paid to investor relation firms, legal counsel and our auditors. We also experienced increased payroll and related costs of $139,491, as well as an increase in our insurance, office and other miscellaneous expenses of $38,827. These increases were offset by reductions in office, travel and depreciation expenses of $22,872 and a decrease in our Delaware franchise taxes of $116,604. Reimbursements from CorLyst totaled $30,751 for rent and other costs during the three months ended March 31, 2021, approximately $6,000 more than reimbursements for the same period in 2020.
We expect the general and administrative expenses to continue to increase as we add staff to support our growing research and development activities and the administration required to operate as a public company.
| 20 |
Interest Expense and Interest Income
Interest expense was $362 and $17,170 for the three months ended March 31, 2021 and 2020, respectively. The interest expense in 2021 was related to our Paycheck Protection Program loan while interest expense in 2020 was related to our $805,000 8% Senior Notes sold in 2019. Included in interest expense is the amortization of debt issuance costs totaling $1,070 for the three months ended March 31, 2020.
Interest income was $4,940 and $829 for the three months ended March 31, 2021 and 2020, respectively. Interest income represents interest earned on money market funds.
Income Tax Benefit.
An income tax benefit of $89,248 was recognized for the three months ended March 31, 2021 as a result of our recording and amortizing the deferred tax liability created in connection with our acquisition of CoNCERT’s license and “Know-How” in exchange for Processa stock that had been issued in the Internal Revenue Code Section 351 transaction on March 19, 2018. The Section 351 transaction treated the acquisition of the Know-How for stock as a tax-free exchange. As a result, under ASC 740-10-25-51 Income Taxes, Processa recorded a deferred tax liability of $3,037,147 for the acquired temporary difference between the financial reporting basis of $11,038,929 and the tax basis of $1,782. The deferred tax liability will be reduced for the effect of the non-deductibility of the amortization of the intangible asset and may be offset by the deferred tax assets resulting from net operating tax losses. This offset results in the recognition of a deferred tax benefit shown in the consolidated statements of operations.
Cash Flows
The following table sets forth our sources and uses of cash and cash equivalents for the three months ended March 31, 2021 and 2020:
| Three months ended | ||||||||
| March 31, | ||||||||
| 2021 | 2020 | |||||||
| Net cash (used in) provided by: | ||||||||
| Operating activities | $ | (2,243,529 | ) | $ | (546,453 | ) | ||
| Financing activities | 9,875,550 | (2,806 | ) | |||||
| Net increase (decrease) in cash | $ | 7,632,021 | $ | (549,259 | ) | |||
Net cash used in operating activities
We used net cash in our operating activities of $2,243,529 and $546,453 during the three months ended March 31, 2021 and 2020, respectively. The increase in cash used in operating activities during the first quarter of 2021 compared to the comparable period in 2020 was primarily related to costs we incurred related to commencing our Phase 2B clinical study for PCS499, Phase 1B clinical study for PCS6422, pre-clinical costs for PCS12852, and professional fees and increased salaries. As part of beginning these clinical trials, we prepaid a portion of certain trial related costs, causing our prepaid expenses to increase by $557,137 during the three months ended March 31, 2021. Our net loss for the three months ended March 31, 2021 of $2,099,481 was $1,225,145 greater than the comparable period in 2020.
As we start the patient enrollment phase of our clinical trials for PCS499 and PCS6422, prepare to submit an IND for PCS12852, and continue to evaluate the other drugs in our portfolio, we anticipate our research and development efforts and on-going general and administrative costs will continue to generate negative cash flows from operating activities for the foreseeable future and these amounts are expected to increase in the future.
Net cash (used in) provided by financing activities
Net cash provided by financing activities during the three months ended March 31, 2021 of $9,875,550 was received in the private placement transaction completed on February 24, 2021, net of cash transaction costs of $363,223. We used net cash in our financing activities of $2,806 related to our underwritten public offering during the three months ended March 31, 2020.
| 21 |
Liquidity
On February 24, 2021, we closed a private placement for the sale of 1,321,132 shares of our common stock at a purchase price of $7.75 per share to accredited and institutional investors for gross proceeds of $10.2 million. Net proceeds from the offering were $9.9 million. We also completed an underwritten public offering in late 2020 where we raised net proceeds from the offering of approximately $17.1 million. As a result of these offerings, at March 31, 2021 we had $23,048,245 in cash.
We have incurred losses and net cash used in our operating activities during the three months ended March 31, 2021, which we expect to continue for the foreseeable future. We do not currently or have since our inception had any sales. We have incurred losses since our inception, devoting substantially all of our efforts toward research and development, and have an accumulated deficit of approximately $27.5 million at March 31, 2021. During the three months ended March 31, 2021, we generated a net loss of approximately $2.1 million. However, we believe our cash balance at March 31, 2021 is adequate to fund our budgeted operations through 2023. Our ability to execute our longer-term operating plans, including unplanned future clinical trials for our portfolio of drugs depend on our ability to obtain additional funding from the sale of equity and/or debt securities, a strategic transaction or other funding transactions. We plan to continue to actively pursue financing alternatives, but there can be no assurance that we will obtain the necessary funding in the future when necessary.
Our estimate of future cash needs is based on assumptions that may prove to be wrong, and we could utilize our available cash sooner than we currently expect. Our ultimate success depends on the outcome of our planned clinical trials and our research and development activities, as disclosed above. We expect to incur additional losses in the future, and we anticipate the need to raise additional capital to fully implement our business plan if the cost of our planned clinical trials are greater than we expect, or they take longer than anticipated. We also expect to incur increased general and administrative expenses in the future due in part to planned increased research and development activities as we conduct a Phase 2B trial for PCS499, a Phase 1B trial for PCS6422, and a Phase 2A clinical trial for PCS12852. In addition, there may be costs we incur as we develop these drug products that we do not currently anticipate requiring us to need additional capital sooner than currently expected.
Our future capital requirements will depend on many factors, including:
| ● | the cost of clinical trials for PCS499, PCS6422 and PCS12852, and the cost of third-party manufacturing; | |
| ● | the delays in patient enrollment due to the COVID-19 pandemic; | |
| ● | the initiation, progress, timing, costs and results of drug discovery, pre-clinical studies, and clinical trials of PCS11T and any other future product candidates; | |
| ● | the number and characteristics of product candidates that we pursue; | |
| ● | the outcome, timing, and costs of seeking regulatory approvals; | |
| ● | the costs associated with hiring additional personnel and consultants as our pre-clinical and clinical activities increase; | |
| ● | the emergence of competing therapies and other adverse market developments; | |
| ● | the costs involved in preparing, filing, prosecuting, maintaining, expanding, defending, and enforcing patent claims, including litigation costs and the outcome of such litigation; | |
| ● | the extent to which we in-license or acquire other products and technologies; and | |
| ● | the costs of operating as a public company. |
| 22 |
Until such time as we can generate substantial product revenues to support our capital requirements, if ever, we expect to finance our cash needs through a combination of public or private equity offerings, debt financings, collaborations and licensing arrangements or other capital sources. To the extent that we raise additional capital through the sale of equity or convertible debt securities, the ownership interest of our stockholders will be or could be diluted, and the terms of these securities may include liquidation or other preferences that adversely affect the rights of our common stockholders.
Contractual Obligations and Commitments
There have been no significant changes to the contractual obligations reported in our Annual Report on Form 10-K for the fiscal year ended December 31, 2020.
Off Balance Sheet Arrangements
At March 31, 2021, we did not have any off-balance sheet arrangements.
Critical Accounting Policies and Use of Estimates
Our discussion and analysis of our financial condition and results of operations are based upon our Unaudited Condensed Consolidated Financial Statements, which have been prepared in accordance with U.S. GAAP. The preparation of these financial statements requires us to make estimates and judgments that affect the reported amounts of assets, liabilities, revenues and expenses and related disclosure of contingent assets and liabilities.
We believe that the estimates, assumptions and judgments involved in the accounting policies described in the “Management’s Discussion and Analysis of Financial Condition and Results of Operations” section of our most recent Annual Report on Form 10-K have the greatest potential impact on our financial statements, so we consider these to be our critical accounting policies. Actual results could differ from the estimates we use in applying our critical accounting policies. We are not currently aware of any reasonably likely events or circumstances that would result in materially different amounts being reported.
There have been no changes in our critical accounting policies from our most recent Annual Report on Form 10-K.
Recently Issued Accounting Pronouncements
We have evaluated recently issued accounting pronouncements and determined that there is no material impact on our financial position or results of operations.
Item 3. Quantitative and Qualitative Disclosures About Market Risk
Item 3 is not applicable to us as a smaller reporting company and has been omitted.
Item 4. Controls and Procedures
Disclosure Controls and Procedures
Our management, with the participation of our Chief Executive Officer (“CEO”) and Chief Financial Officer (“CFO”), evaluated the effectiveness of our disclosure controls and procedures as of the end of the period covered by this Report. Based upon that evaluation, the CEO and CFO concluded that our disclosure controls and procedures as of the end of the period covered by this Report were not effective in providing reasonable assurance in the reliability of our report as of the end of the period covered by this report.
In our 2020 Annual Report on Form 10-K, we identified the following material weaknesses in our internal control over financial reporting, which are common in many small companies with limited staff including: (i) certain entity level controls; (ii) inadequate segregation of duties throughout the entire year; and (iii) insufficient documentation of certain policies and procedures for transaction processing, accounting and financial reporting with respect to the requirements and application of both GAAP and SEC guidelines, their related controls and the operation thereof. These material weaknesses continue to be present at March 31, 2021.
| 23 |
Changes in Internal Control over Financial Reporting
During our quarter ended March 31, 2021, we implemented changes to our cash disbursements in order to strengthen our internal controls. Changes include a central accounts payable email address for invoice intake and processing, and an electronic invoice approval process. We also added additional approval requirements for wire transfers over certain amounts. We believe these additions have materially improved our internal control over financial reporting. We are continuing to take remediation actions to rectify our control deficiencies (including material weaknesses) through the adoption and implementation of written policies and procedures for transaction processing, accounting and financial reporting, as well as strengthening our supervisory review processes.
In designing and evaluating the disclosure controls and procedures, management recognizes that any controls and procedures, no matter how well designed and operated, can provide only reasonable assurance of achieving the desired control objectives. In addition, the design of disclosure controls and procedures must reflect the fact that there are resource constraints, and that management is required to apply judgment in evaluating the benefits of possible controls and procedures relative to their costs.
We are currently not a party to any material legal proceedings.
There have been no material changes to our risk factors as described in Item 1A of our Annual Report on Form 10-K for the year ended December 31, 2020.
Item 2. Unregistered Sales of Equity Securities and Use of Proceeds
(a) Recent Sale of Unregistered Securities
On February 24, 2021, we closed a private placement for the sale of 1,321,132 shares of our common stock at a purchase price of $7.75 per share to accredited and institutional investors for gross proceeds of $10.2 million. Net proceeds from the offering were $9.9 million. Following closing the offering, we filed a Form S-3 for the selling shareholders of these shares, which was declared effective by the U.S. Securities and Exchange Commission on April 12, 2021.
(b) Use of Proceeds from Public Offering of Common Stock
None.
(c) Issuer Purchases of Equity Securities
We did not repurchase any shares of our common stock during the three months ended March 31, 2021.
Item 3. Defaults Upon Senior Securities
None.
Item 4. Mine Safety Disclosures
Not applicable.
None.
| SEC Ref. No. | Title of Document | |
| 31.1* | Rule 153-14(a) Certification by Principal Executive Officer | |
| 31.2* | Rule 153-14(a) Certification by Principal Financial Officer | |
| 32.1*++ | Section 1350 Certification of Principal Executive Officer and Principal Financial Officer | |
| 99.1 | XBRL Files |
* Filed herewith.
++ This certification is being furnished solely to accompany this Quarterly Report pursuant to 18 U.S.C. Section 1350 and are not being filed for purposes of Section 18 of the Securities Exchange Act of 1934, as amended, and are not to be incorporated by reference into any filing of the Company, whether made before or after the date hereof, regardless of any general incorporation language in such filing herewith.
| 24 |
SIGNATURES
Pursuant to the requirements of the Securities Exchange Act of 1934, the registrant has duly caused this report to be signed on its behalf by the undersigned thereunto duly authorized.
| PROCESSA PHARMACEUTICALS, INC. | ||
| By: | /s/ David Young | |
| David Young | ||
Chief Executive Officer (Principal Executive Officer) Dated: May 13, 2021 | ||
| By: | /s/ James Stanker | |
James Stanker Chief Financial Officer (Principal Financial and Accounting Officer) Dated: May 13, 2021 | ||
| 25 |